

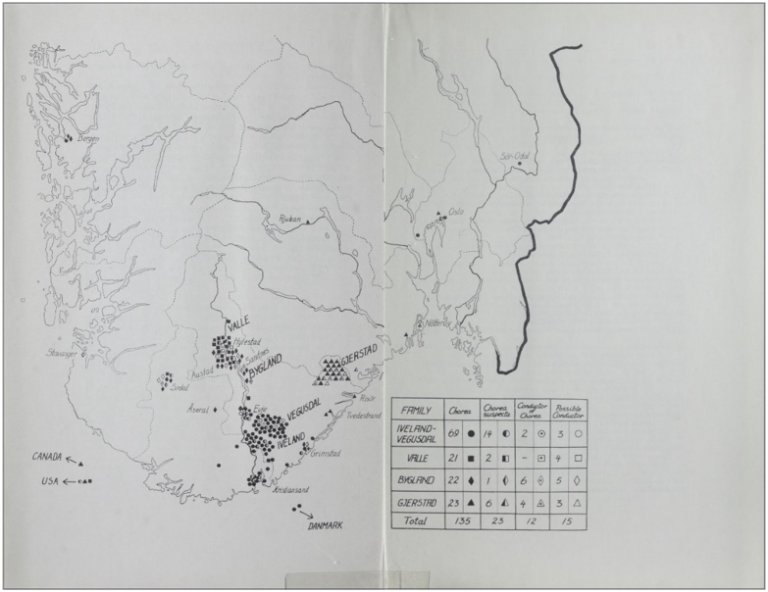
Kartutdrag fra Ørbeck og Quelpruds bok fra 1954
Huntingtons sykdom
Huntingtons sykdom – en medisinsk gåte fra Setesdal. Kan deler av svaret eksistere i Helsearkivregisteret?
Hvem kartla og oppdaget sykdommen Huntingtons? Legen som har fått sykdommen oppkalt etter seg, var en amerikaner ved navn Huntington som beskrev sykdommen i 1872 (1). Men allerede tolv år tidligere (1860) beskriver den nyutdannede norske distrikslengen Johan Christian Lund en arvelig sykdom i sin «Medicinal beretning» fra legedistriktet Setesdal (2). Lund beskriver at sykdommen på folkemunne omtales som «Rykkja», «kjerringrykkja» eller «arvesygen».

Om Huntingtons sykdom
Huntingtons sykdom er en sjelden arvelig hjernesykdom som gradvis fører til motoriske, psykiske og kognitive forverringer inntil pasientens død, 15-20 år etter diagnosen er satt. Symptomdebut er vanligvis ved 35-45 års alder. Det finnes ingen behandling av sykdommen. I Norge anslås det å være mellom 300-400 personer som enhver tid lever med diagnosen, og forekomsten på verdensbasis varier mellom 0,25 til 7 tilfeller pr. 100 000 innbygger (5; 6). Beskrivelser av sykdommen kan spores tilbake til middelalderen, og har en norsk historie som like gjerne kunne endt med at sykdommen het Lunds sykdom i dag (6; 7).
Den tidligste omfattende beskrivelsen av Huntingtons sykdom i Norge stammer fra distriktslege Johan Christian Lunds dokumentasjon om "Sundhetstilstanden i Sætersdalen" fra 1860, der han refererte til sykdommen som "Setesdalsrykkja" (2).
Den amerikanske legen George Huntington ga en mer detaljert vitenskapelig beskrivelse av sykdommen i 1872 (1). Ettersom begge beskrivelsene var såpass nært i tid, ble sykdommen kalt "Huntington-Lund Chorea" i en kort periode. Huntington er den som i senere tid fikk navnet sitt knyttet til sykdommen. I dag er sykdommens offisielle navn i ICD kodeverket «Huntingtons sykdom», men navnet «Setesdalsrykkja» står fortsatt i den norske utgaven (8).
I 1993 ble Huntington-genet med den sykdomsfremkallende mutasjonen kartlagt ved å studere to isolerte landsbyer i Venezuela gjennom Hereditary Disease Foundation. Disse landsbyboerne hadde en uvanlig høy forekomst av Huntingtons sykdom, lignende det som ble observert i relativt isolerte Setesdal omtrent 125 år tidligere (9).
Huntingtons sykdom utvikler seg gradvis over 15-25 år fra diagnosetidspunktet. Ifølge Senter for sjeldne diagnoser ved Oslo Universitetssykehus stilles diagnosen etter grundig utredning basert på symptomer, funn ved nevrologisk undersøkelse og resultatene fra gentester (10).
Fra heksebrenning til genforskning
I dag er Huntingtons sykdom nøye beskrevet både i ICD-kodeverket og på genetisk nivå gjennom Huntington-genet. Dette har imidlertid ikke alltid vært tilfelle. Det finnes kilder som skildrer heksebrenning av kvinner med sykdommen i enda tidligere tider, og i Setesdal er det på 1800-tallet advarsler mot å la seg gifte inn i «rykkje-slekter». I 1946 hevdet flere at sykdommen langt på vei var utryddet i Norge – men dette viste seg å ikke stemme (3; 4). Symptomene på sykdommen har lenge vært kjent blant befolkningen i Setesdal og kan spores tilbake til middelalderen i andre kilder, da symptomene ble omtalt som Sankt Vitus’ dans – ofte også sammenblandet med andre sykdommer med lignende symptomer (7; 3).
Middelalderen
Sankt Vitus (ca. født 290, død 303 e.Kr.) er en kristen barnehelgen som led martyrdøden under keiser Diokletians forfølgelse av kristne. Mens historien om ham er basert på usikre kilder, ble han i middelalderen ansett som en helgen som spesielt kunne hjelpe barn og unge som led av epilepsi og kramper. På grunn av dette og den senmiddelalderske tyske skikken med å danse foran hans statue, har nervesykdommen sanktveitsdans (chorea, fra det greske khoreia, «kordans») fått sitt navn fra ham. Ufrivillige rykninger – chorea - er et symptom som kan oppstå ved flere sykdommer, inkludert Huntingtons sykdom. Derfor har Huntingtons sykdom også blitt referert til som «Huntingtons Chorea» (11; 7).
Hekseforfølgelse
I 1630, i en liten landsby i England ved navn Bures St. Mary, ble flere kvinner anklaget for å være hekser på grunn av bisarr oppførsel og rykninger som lignet på symptomer ved Huntingtons sykdom. Samme år satte tre menn med deres koner fra denne landsbyen kursen til Amerika for å bosette seg langs den østlige kysten. Disse mennene var brødre, og minst syv av deres kvinnelige etterkommere ble senere anklaget for hekseri og stilt for retten.
Moderne tid
I løpet av 1800-tallet ble sykdommen stadig grundigere beskrevet. Charles Waters publiserte sine observasjoner i Dunglisons "Practice of Medicine" i 1842, noe som generelt regnes som den første skriftlige beretningen om sykdommen. Imidlertid var det med Lunds utredning i 1860 at sykdommen ble grundigere utforsket, med et detaljert familietre som strakte seg over fire generasjoner, og som dokumenterte nedarvede tilfeller av den samme sykdommen i Setesdal (2).

Dr. Huntington
George Huntington ble født inn i en familie av allmennleger i East Hampton, New York, USA, i 1850. Han gjennomførte sin medisinske utdanning ved College of Physicians and Surgeons of Columbia University og ble uteksaminert i 1871. Etter å ha returnert til sitt hjemsted begynte han å praktisere medisin og rapporterte raskt om tilfeller av demens og korea hos middelaldrende individer. Disse tilfellene fulgte et mønster med familiær opprinnelse og overføring gjennom autosomal (kjønnsuavhengig) dominant arv. Han dokumenterte også lignende tilfeller som tidligere var blitt undersøkt av hans far og bestefar, og han studerte disse familiene i stor detalj; farens notater er tydelige i Huntingtons originale manuskript.
De familiene han studerte var etterkommere av en Jeffrey Francis som emigrerte fra England til Long Island i 1634, og det antydes at genet ble overført med ham. Bare 20 år gammel, den 15. februar 1872, presenterte Huntington sine observasjoner med tittelen "On Chorea" for Meigs og Mason Academy of Medicine i Middleport, Ohio, USA. Denne rapporten ble publisert i "Medical and Surgical Reporter" senere samme år og regnes som den første vitenskapelige publikasjonen om Huntingtons sykdom (1).

Selv om tidligere forskere hadde rapportert om denne tilstanden, var det særegenheten og klarheten i Huntingtons beskrivelse som førte til at han ble tilknyttet eponymet "Huntingtons sykdom", iblant forkortet til HD. Gjennom arbeidet til Huntington fikk de sykdomsrammede endelig menneskeligheten sin tilbake, etter å ha vært til spott og spe gjennom uminnelige tider.
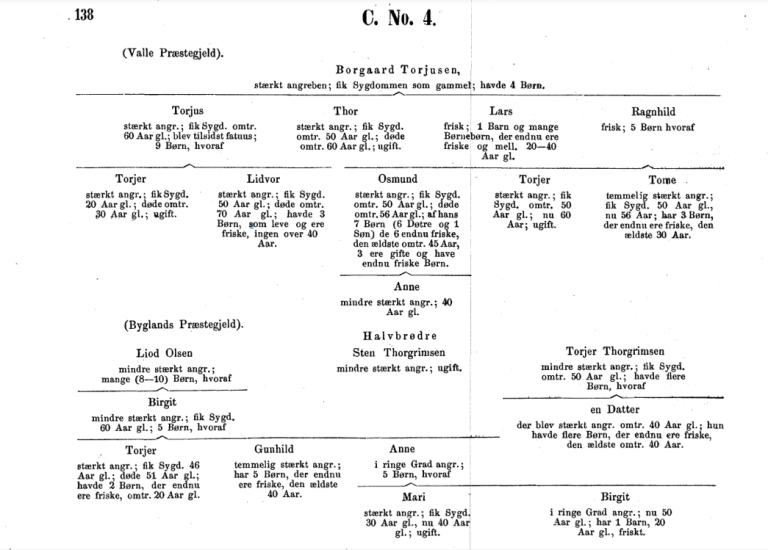
Setesdalsrykkja
Det har også vært senere publikasjoner om Hutingtons sykdom i Norge. En av de mer omfattende er for eksempel Alf Ørbeck og Thordar Quelpruds bok «Setesdalsrykkja» fra 1954.
I Ørbeck og Quelpruds bok fra 1954 fremholder de at Huntingtons sykdom i Setesdal og tilliggende bygder vært kjent i uminnelige tider, bare under navnet "rykkja". Folkemeningen i bygda antydet for lenge siden at "rykkja" var arvelig, og i 1820-årene advarte presten i Vegusdal fra prekestolen mot å gifte seg inn i "rykkeslekter". "Rykkeslekter" er blitt stigmatisert for å ha en vanærende sykdom, og i folkemeningen i bygda skal det ha vært en foraktelig tone da bygdefolket undret seg over de som ønsket å gifte seg inn i en slik "rykkeslekt".



Forfatterne argumenterer for at sykdommen også bør bære distriktslege Johan Christian Lunds navn, siden han beskrev sykdommen 12 år før Huntington publiserte sine funn.
Ørbeck og Quelprud tar også med et sagn om hvordan "Setesdalrykka" oppsto.
Kartutdrag fra Ørbeck og Quelpruds viser kjent forekomst og geografisk spredning av chorea hereditaria i Setesdal og omegn. Legg også merke til pilene som peker på utvandrede tilfeller av sykdommen, til USA og Canada.
Feilfoldede proteiner - Huntington-genet kartlegges
Fra 1979 har Hereditary Disease Foundation bidratt til et amerikansk-venezuelansk forskningsprosjekt. Et team bestående av fremstående internasjonale leger og helseforskere gjorde mange reiser til rurale fiskelandsbyer langs breddene av Lake Maracaibo i Venezuela. Disse landsbyene har den høyeste konsentrasjonen av Huntingtons sykdom i verden, og huser den største familien som lever med sykdommen. Grunnleggeren av denne familien levde på begynnelsen av 1800-tallet. Slektstreet hennes omfatter over 18 000 individer fordelt over 10 generasjoner. Mange av disse er enten berørt av sykdommen eller i risikogruppen for å utvikle den (13). De isolerte venezuelanske landsbyene minner dermed mye om de avsidesliggende områdene i Setesdal, hvor familier med sykdomsrammede individer ble beskrevet av Lund i 1860.
Huntingtons i dag
Mellom 300 og 400 personer anslås å leve med Huntingtons i Norge i dag, og om lag 1000 anslås å være i risiko (10). Prevalensen på verdensbasis varierer mellom verdensdeler. Den varierer fra laveste forekomst i asiatiske og afrikanske deler av verden med 0,25-2 tilfeller pr 100 000 innbygger, til høyest forekomst i nord-amerikansk og europeisk befolkning med en forekomst på mellom 3-7 pr 100 000 innbygger (5).
Genet som koder for sykdommen ble kartlagt i 1993 (12), men over 30 år senere finnes det fortsatt ingen behandling. Helsetjenesten har dermed fått en rolle som en genetisk veileder til enkeltindivider og familier, ved siden av å tilrettelegge for et best mulig liv med sykdommen for de som rammes (14).
Den arvelige disponeringen er lik for kvinner og menn, og dersom en som bærer genet får barn er det 50% risiko for at barnet arver genet. Denne nedarvingen kalles autosomal dominant, og betyr også at genet ikke kan «hoppe over» generasjoner (10).
De senere årene er gentesting blitt tilgjengelig for de med arvelig risiko for sykdommen, men dette oppleves av mange som en svært belastende prosess, og ifølge en norsk studie så velger så mange som 80 % å ikke gjennomføre testen (15). Genetisk veiledning er tilgjengelig for gravide og risikopersoner som planlegger å få barn, men lovverket er under stadig utvikling og diskusjon (16; 17).
Huntingtons sykdom er en sjelden, men alvorlig og svært belastende diagnose for de pasientene og familiene som rammes. I dag spiller flere betydelige aktører en rolle innenfor Huntingtons sykdom i Norge og globalt. Blant disse er Senter for sjeldne sykdommer ved Oslo Universitetssykehus, Frambu kompetansesenter for sjeldne diagnoser, Landsforeningen for Huntingtons sykdom, og de fem regionale ressurssentrene i landet. Ressurssentrene har en viktig rolle som pådrivere for kunnskapsbasert praksis for pasientgruppen, og de jobber for å sikre kvalitet i helsetjenesten og for å oppnå ny kunnskap om sykdommen. Arv og genetikk er avgjørende for å løse gåten rundt sykdommen og sikre pasientene og deres familier utsikter til bedring (10; 18; 19; 14).
Huntingtons sykdom i Helsearkivregisteret
Helsearkivregisteret (HAREG) har som formål å sikre, bevare og gjøre helsedata fra spesialisthelsetjenesten tilgjengelig for forskning. For sjeldne diagnoser kan det ofte være krevende å finne nok individer til tilfredsstillende studieutvalg. Med stadig mer helsedata og pasientjournaler i Helsearkivregistret kan denne helsedatakilden bli et viktig bidrag til forskning på sjeldne sykdommer for små pasientgrupper. På mange måter åpner Helsearkivregisteret en ny dimensjon, fordi vi kan levere forskningsdata som strekker seg langt bakover i tid, noe som tidligere ikke har vært mulig.
De viktigste medisinske metadataene i HAREG er diagnosene som registreres fra pasientjournalene. Der innleggelsesdiagnosene er kodet etter det internasjonale diagnosekodeverket ICD, benyttes dette kodeverket og koden. Dersom kodeverk ikke er brukt av klinikeren, registreres diagnosen som en fritekstvariabel. Tendensen er klar, jo eldre journalmateriale – jo mer ukodede diagnoser. Derfor er det ikke alltid like enkelt å gjøre gode spørringer, dessuten er de tidlige versjonene av ICD-kodeverket mindre presise enn dagens.
Huntingtons statistikk fra Helsearkivregisteret
I statistikken fra HAREG for Huntingtons sykdom har vi gjort spørringer på journaler med ICD-kodene for Huntington i versjon 8-10, samt en fritekstspørring på diagnoser for innhold «Huntington».
Alt i alt er det 713 unike registrerte diagnoser som samsvarer med Huntingtons sykdom i HAREG. Fordi diagnosen kan være registrert flere ganger i samme journal, for eksempel ved gjentatte innleggelser, blir antallet unike diagnoser pr. journal 343. Teller vi igjen journaler med en av Huntington diagnosene registrert lander vi på 291 unike journaler.
Fra HAREG hentet vi ut 291 journaloppføringer, og etter datavask endte utvalget på 255 journaloppføringer som er inkludert i videre analyser. Ved å fjerne de uten komplett fødselsdato/dødsdato endte vi med 242 journaler fordelt på 228 unike personer (14 personer har to journaler).
Antall | Gjennomsnitt alder død | Gj.snitt alder første diagnose | Gj.snitt leveår med diagnosen | |
Kvinner | 118 | 62 | 54 | 9 |
Menn | 124 | 62 | 56 | 6 |
Totalt | 242 | 62 | 55 | 7,4 |
| Min | Max | Mean | Std |
Alder død | 18 | 97 | 62,3 | 14,6 |
Alder første diagnose | 16 | 96 | 55 | 15,3 |
Leveår med diagnosen | 0 | 30 | 7,4 | 5,9 |
For Huntingtonutvalget i HAREG er det gjort beregninger på gjennomsnittsalder for første diagnosedato i journalene som finnes i HAREG, fordelt på kjønn. Det er videre sett på alder ved død og levealder med diagnosen.
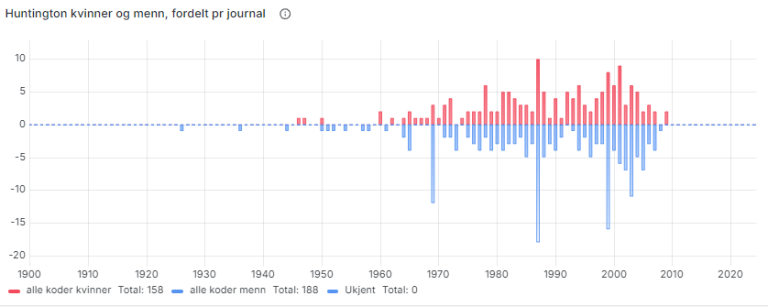
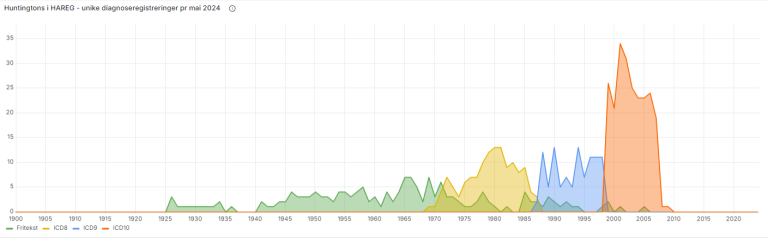
Helsearkivregisterets ustrukturerte Huntingtons data
I tillegg til diagnoser som medisinske metadata består HAREG av enorme mengder ustrukturert journalinnhold. For deler av registeret brukes kunstig intelligens til å definere språkkonsepter som kan gi innsikt om innholdet, uavhengig av metadata. Friteksten fra de digitaliserte journalene blir maskinlest, og der kvaliteten er god nok er dette en uvurderlig kilde for supplerende statistikk og innsikt i HAREG.
For Huntingtons sykdom har vi brukt teknologien til å definere språkkonsepter for diagnosen og symptomer. Dette blir så til komplekse søk, hvor ordene i hvert språkkonsept brukes til å definere utvalg med data hvor ord fra konseptet er til stede.
Tidslinje for Huntingtons sykdom
Klikk her for å åpne tidslinjen i ny fane.




Ønsker du å vite mer?
Vi ønsker at våre data skal brukes! Ta gjerne kontakt med oss hvis du vil vite mer om innholdet i HAREG. Du er velkommen til oss, og vi kommer gjerne til ditt fagmiljø for å presentere innholdet og mulighetene for forskning som ligger i Helsearkivregisteret.
helsearkivregisteret@arkivverket.no
https://helsedata.no/no/forvaltere/arkivverket/helsearkivregisteret
Referanser
1. Huntington, George. On Chorea. The Medical and Surgical Reporter. 15, 1872, Vol. 26, 789.
2. Lund, Johan Christian. Medicinal Beretning for Sætesdal Lægedistrikt. 1860.
3. Ørbeck, Alf og Quelprud, Thordar. Setesdalsrykka - Chorea Progressiva Hereditaria. s.l. : Det Norske Vitenskapsakademi, 1954.
4. Lie, Anne Kvein. Setesdalsrykkja - 100 år etter. Tidsskrif for den Norsk legeforening. 2035, 2008, 128.
5. The Haymarket Medical Network. Rare Disease Advisor. Huntington Disease (HD). [Online] 9 8, 2023. [Cited: 3 14, 2024.] https://www.rarediseaseadvisor.com/disease-info-pages/huntington-disease-epidemiology/.
6. Store norske leksikon. Store medisinske leksikon. Huntingtons sykdom. [Online] 3 8, 2024. [Cited: 3 12, 2024.] https://sml.snl.no/Huntingtons_sykdom.
7. Heiberig, Arvid. Huntingtons sykdom. Tidsskrift for den Norske legeforening. 2214-7, 2008, 128.
8. Verdens helseorganisasjon / Direktoratet for e-helse. ICD - 10 . Den internasjonale statistiske klassifikasjon av sykdommer og beslektede helseproblemer. [Online] 1 1, 2024. [Cited: 3 12, 2024.] https://finnkode.ehelse.no/#icd10/0/0/0/2599590.
9. Hereditary Disease Foundation. Huntington´s Disease in Venezuela. [Online] https://www.hdfoundation.org/venezuela.
10. Oslo Universitetssykehus. Senter for sjeldne diagnoser. Diagnoseinformasjon Huntingtons sykdom. [Online] 6 9, 2023. [Cited: 3 12, 2024.] https://www.oslo-universitetssykehus.no/fag-og-forskning/nasjonale-og-regionale-tjenester/senter-for-sjeldne-diagnoser/diagnoseinformasjon-fra-senter-for-sjeldne-diagnoser/huntingtons-sykdom-#huntingtons-sykdom-pa-5-minutter-for-fastleger.
11. Wikipedia. Wikipedia - English. Saint Vitus. [Online] 10 1, 2023. [Cited: 3 14, 2024.] https://en.wikipedia.org/wiki/Saint_Vitus.
12. MacDonald, Marcy E. , et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Cell. 1993, Vol. 72, 6.
13. Hereditary Disease Foundation . hdfoundation.org. Huntington's Disease in Venezuela. [Online] [Cited: 3 14, 2024.] https://www.hdfoundation.org/venezuela.
14. Frambu kompetansesenter for sjeldne diagnoser. frambu.no. Om oss. [Online] [Cited: 3 14, 2024.] https://frambu.no/om-oss/.
15. Tillerås, Kristine H. et al. Psychological reactions to predictive genetic testing for Huntington's disease: A qualitative study. Journal of Genetic Counseling. 2020.
16. Regjeringen. regjeringen.no. Høring – forslag til endringer i bioteknologilovens regler for preimplantasjonsdiagnostikk (PGD) og forbud mot genetisk testing av barn utenfor helsetjenesten. [Online] 2 28, 2023. [Cited: 3 14, 2024.] https://www.regjeringen.no/no/dokumenter/horing-forslag-til-endringer-i-bioteknologilovens-regler-for-preimplantasjonsdiagnostikk-pgd-og-forbud-mot-genetisk-testing-av-barn-utenfor-helsetjenesten/id2964596/?expand=horingssvar.
17. Landsforeningen for Huntingtons sykdom i samarbeid med Senter for sjeldne diagnoser. oslo-universitetssykehus.no. Et vanskelig valg Huntingtons sykdom Informasjon om presymptomatisk test. [Online] 10 2019. [Cited: 3 14, 2024.] https://www.oslo-universitetssykehus.no/4a2078/contentassets/6a14f1ade8f147b2919c170cd7fd9efa/dokumenter/huntingtons-sykdom/et-vanskelig-valg_hs_19.pdf.
18. Landsforeningen for Huntingtons sykdom. Om oss. [Online] [Cited: 3 14, 2024.] https://www.huntington.no/?k=4529.
19. Fagnettverk Huntington. [Online] [Cited: 3 14, 2024.] https://fagnettverkhuntington.no/index.php/om-fagnettverket.